publications
2025
-
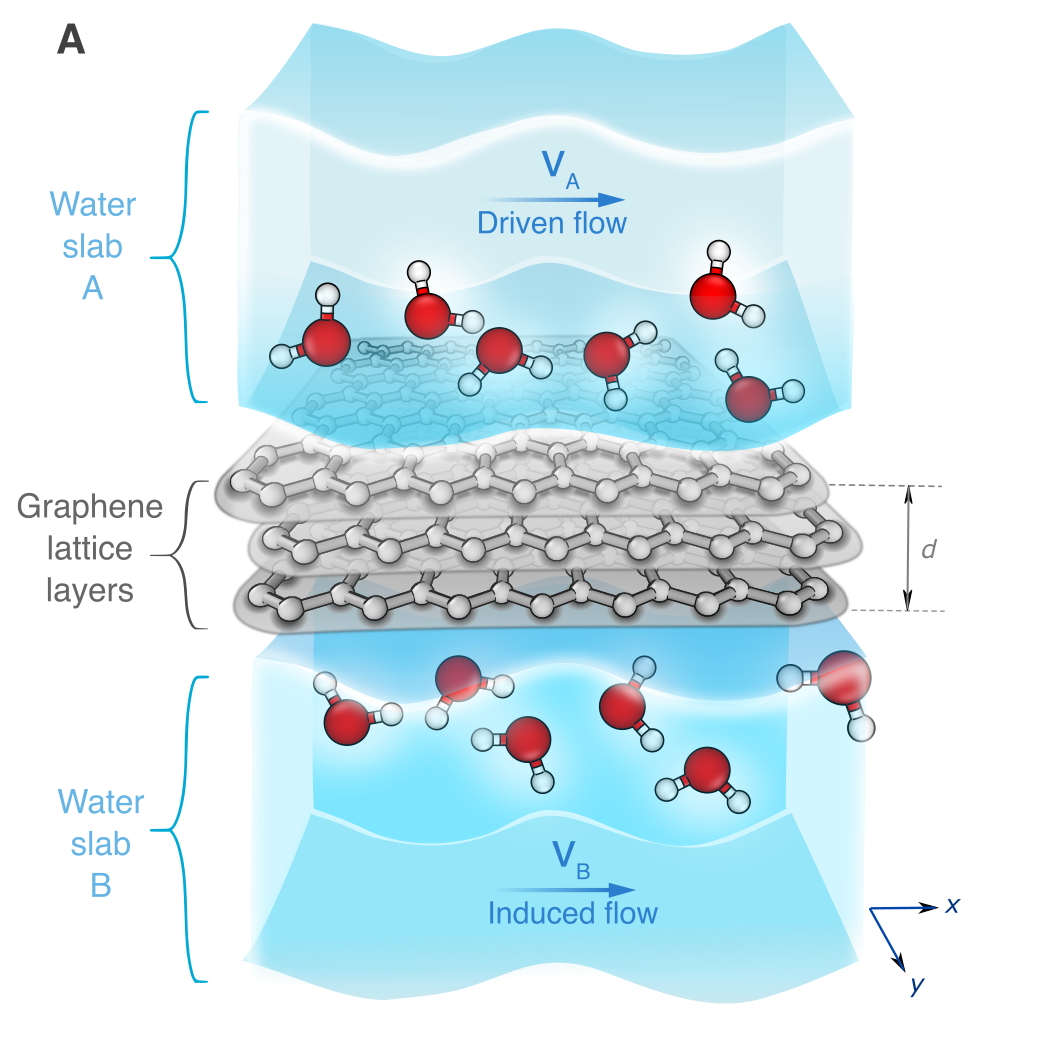 Momentum tunnelling between nanoscale liquid flowsBaptiste Coquinot, Anna T. Bui, Damien Toquer, Angelos Michaelides, Nikita Kavokine, Stephen J. Cox, and Lydéric BocquetNature Nanotechnology, Jan 2025
Momentum tunnelling between nanoscale liquid flowsBaptiste Coquinot, Anna T. Bui, Damien Toquer, Angelos Michaelides, Nikita Kavokine, Stephen J. Cox, and Lydéric BocquetNature Nanotechnology, Jan 2025The world of nanoscales in fluidics is the frontier where the continuum of fluid mechanics meets the atomic, and even quantum, nature of matter. While water dynamics remains largely classical under extreme confinement, several experiments have recently reported coupling between water transport and the electronic degrees of freedom of the confining materials. This avenue prompts us to reconsider nanoscale hydrodynamic flows under the perspective of interacting excitations, akin to condensed matter frameworks. Here we show, using a combination of many-body theory and molecular simulations, that the flow of a liquid can induce the flow of another liquid behind a separating wall, at odds with the prediction of continuum hydrodynamics. We further show that the range of this ‘flow tunnelling’ can be tuned through the solid’s electronic excitations, with a maximum occurring when these are at resonance with the liquid’s charge density fluctuations. Flow tunnelling is expected to play a role in global transport across nanoscale fluidic networks, such as lamellar graphene oxide or MXene membranes. It further suggests exploiting the electronic properties of the confining walls for manipulating liquids via their dielectric spectra, beyond the nature and characteristics of individual molecules.
@article{Coquinot2025, author = {Coquinot, Baptiste and Bui, Anna T. and Toquer, Damien and Michaelides, Angelos and Kavokine, Nikita and Cox, Stephen J. and Bocquet, Lyd{\'e}ric}, title = {Momentum tunnelling between nanoscale liquid flows}, journal = {Nature Nanotechnology}, year = {2025}, month = jan, day = {02}, issn = {1748-3395}, doi = {10.1038/s41565-024-01842-8}, url = {https://doi.org/10.1038/s41565-024-01842-8}, publisher = {Nature Portfolio}, } -
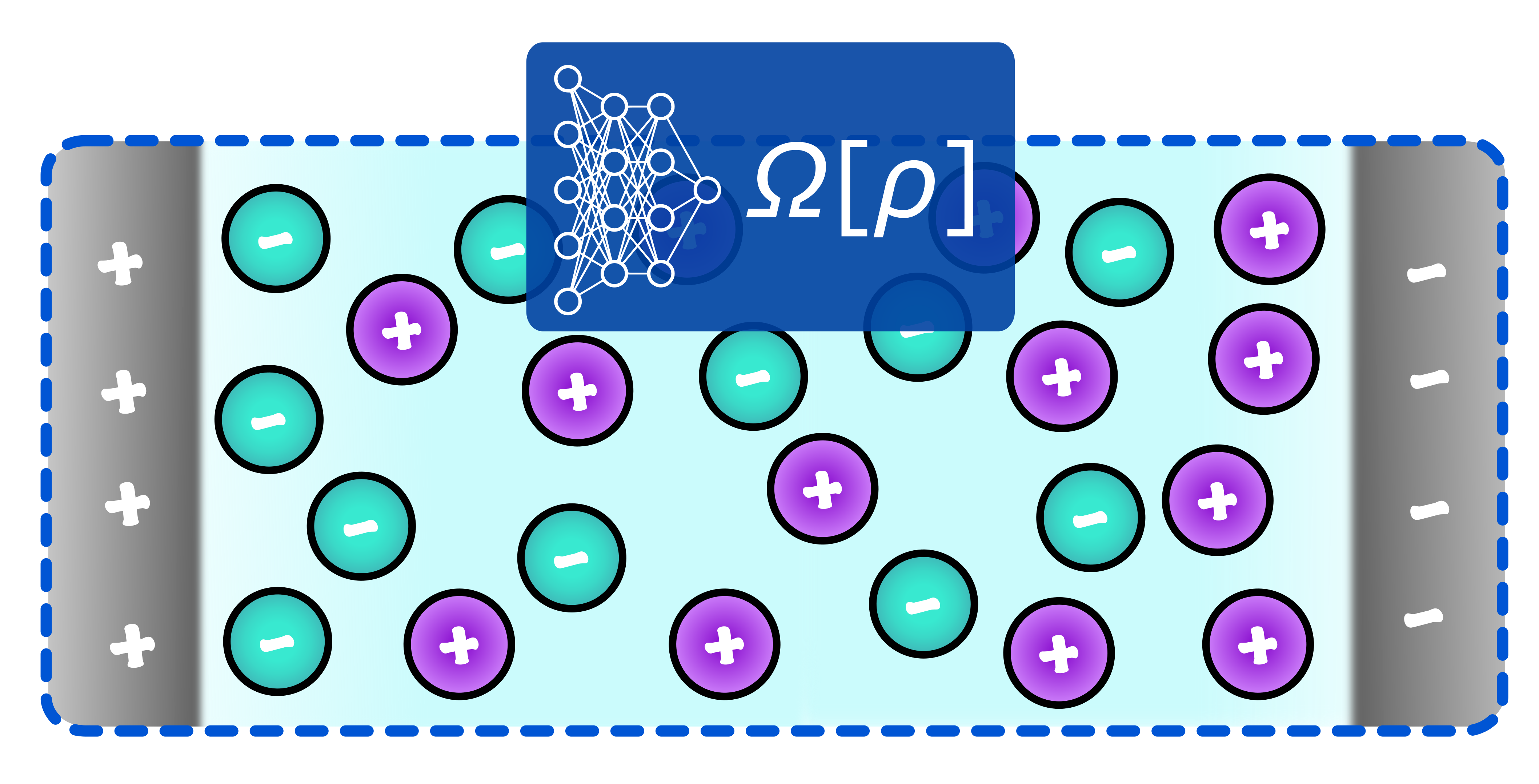 Learning classical density functionals for ionic fluidsAnna T. Bui, and Stephen J. CoxPhys. Rev. Lett., Apr 2025
Learning classical density functionals for ionic fluidsAnna T. Bui, and Stephen J. CoxPhys. Rev. Lett., Apr 2025Accurate and efficient theoretical techniques for describing ionic fluids are highly desirable for many applications across the physical, biological and materials sciences. With a rigorous statistical mechanical foundation, classical density functional theory (cDFT) is an appealing approach, but the competition between strong Coulombic interactions and steric repulsion limits the accuracy of current approximate functionals. Here, we extend a recently presented machine learning (ML) approach [Sammüller et al., Proc. Natl. Acad. Sci. USA, 120, e2312484120 (2023)] designed for systems with short-ranged interactions to ionic fluids. By adopting ideas from local molecular field theory, the framework we present amounts to using neural networks to learn the local relationship between the one-body direct correlation functions and inhomogeneous density profiles for a "mimic” short-ranged system, with effects of long-ranged interactions accounted for in a mean-field, yet well-controlled, manner. By comparing to results from molecular simulations, we show that our approach accurately describes the structure and thermodynamics of the prototypical model for electrolyte solutions and ionic liquids: the restricted primitive model. The framework we present acts as an important step toward extending ML approaches for cDFT to systems with accurate interatomic potentials.
@article{bui2025learningclassicaldensityfunctionals, author = {Bui, Anna T. and Cox, Stephen J.}, title = {Learning classical density functionals for ionic fluids}, journal = {Phys. Rev. Lett.}, volume = {134}, issue = {14}, pages = {148001}, numpages = {8}, year = {2025}, month = apr, publisher = {American Physical Society}, doi = {10.1103/PhysRevLett.134.148001}, url = {https://link.aps.org/doi/10.1103/PhysRevLett.134.148001}, } - arXiv
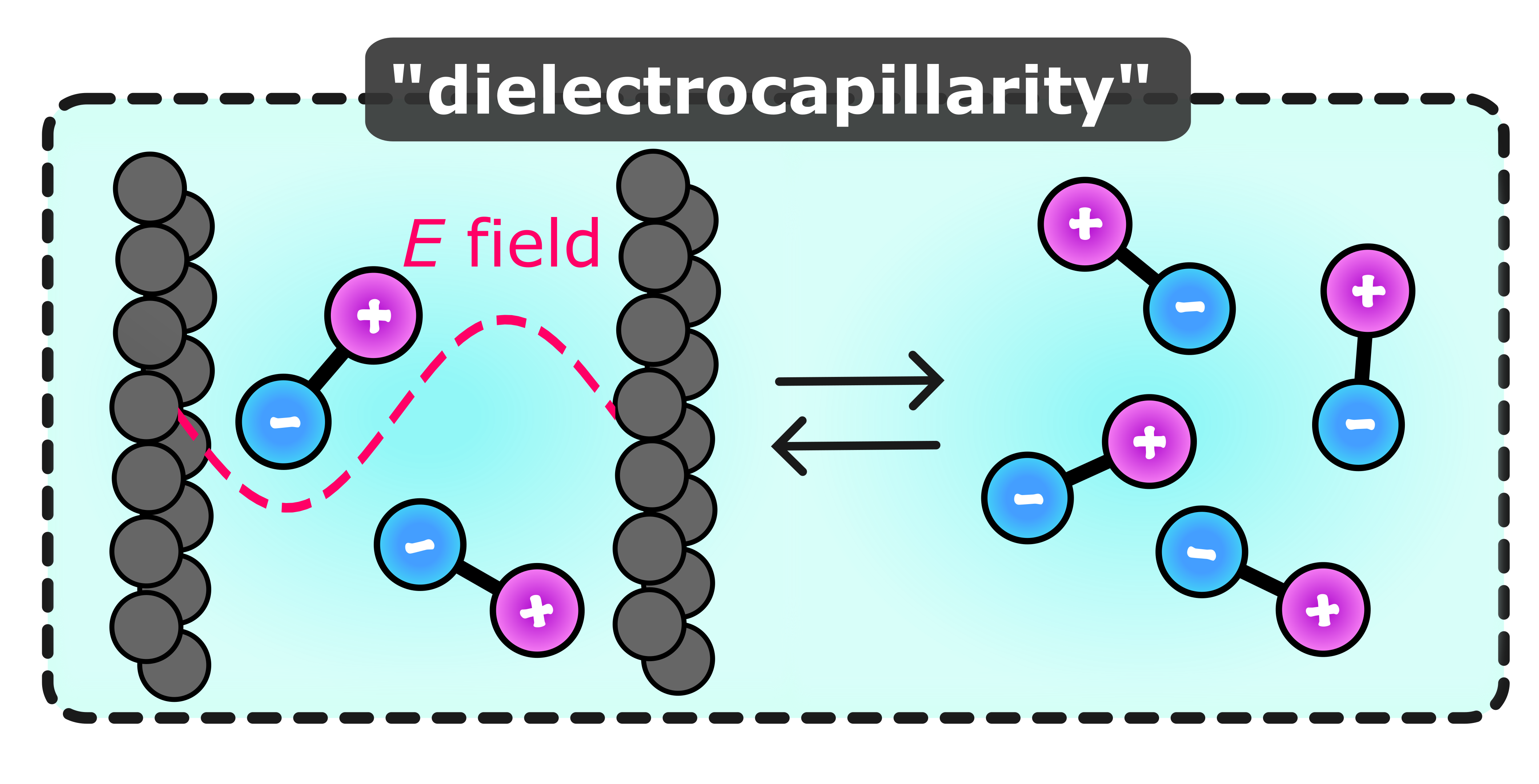 Dielectrocapillarity for exquisite control of fluidsAnna T. Bui, and Stephen J. CoxApr 2025
Dielectrocapillarity for exquisite control of fluidsAnna T. Bui, and Stephen J. CoxApr 2025Spatially varying electric fields are prevalent throughout nature and technology, arising from heterogeneity inherent to all physical systems. Inhomogeneous electric fields can originate naturally, such as in nanoporous materials and biological membranes, or be engineered, e.g., with patterned electrodes or layered van der Waals heterostructures. While uniform fields cause free ions to migrate, for polar fluids they simply act to reorient the constituent molecules. In contrast, electric field gradients (EFGs) induce a dielectrophoretic force, offering exquisite electrokinetic control of a fluid, even in the absence of free charge carriers. EFGs, therefore, offer vast potential for optimizing fluid behavior under confinement, such as in nanoporous electrodes, nanofluidic devices, and chemical separation materials. Yet, EFGs remain largely unexplored at the microscopic level owing to the absence of a rigorous, first principles theoretical treatment of electrostrictive effects. By integrating state-of-the-art advances in liquid state theory and deep learning, we reveal how EFGs modulate fluid structure and capillary phenomena. We demonstrate, from first principles, that dielectrophoretic coupling enables tunable control over the liquid-gas phase transition, capillary condensation, and fluid uptake into porous media. Our findings establish "dielectrocapillarity” – the use of EFGs to control confined fluids – as a powerful tool for controlling volumetric capacity in nanopores, which holds immense potential for optimizing energy storage in supercapacitors, selective gas separation, and tunable hysteresis in neuromorphic nanofluidic devices.
- IOP
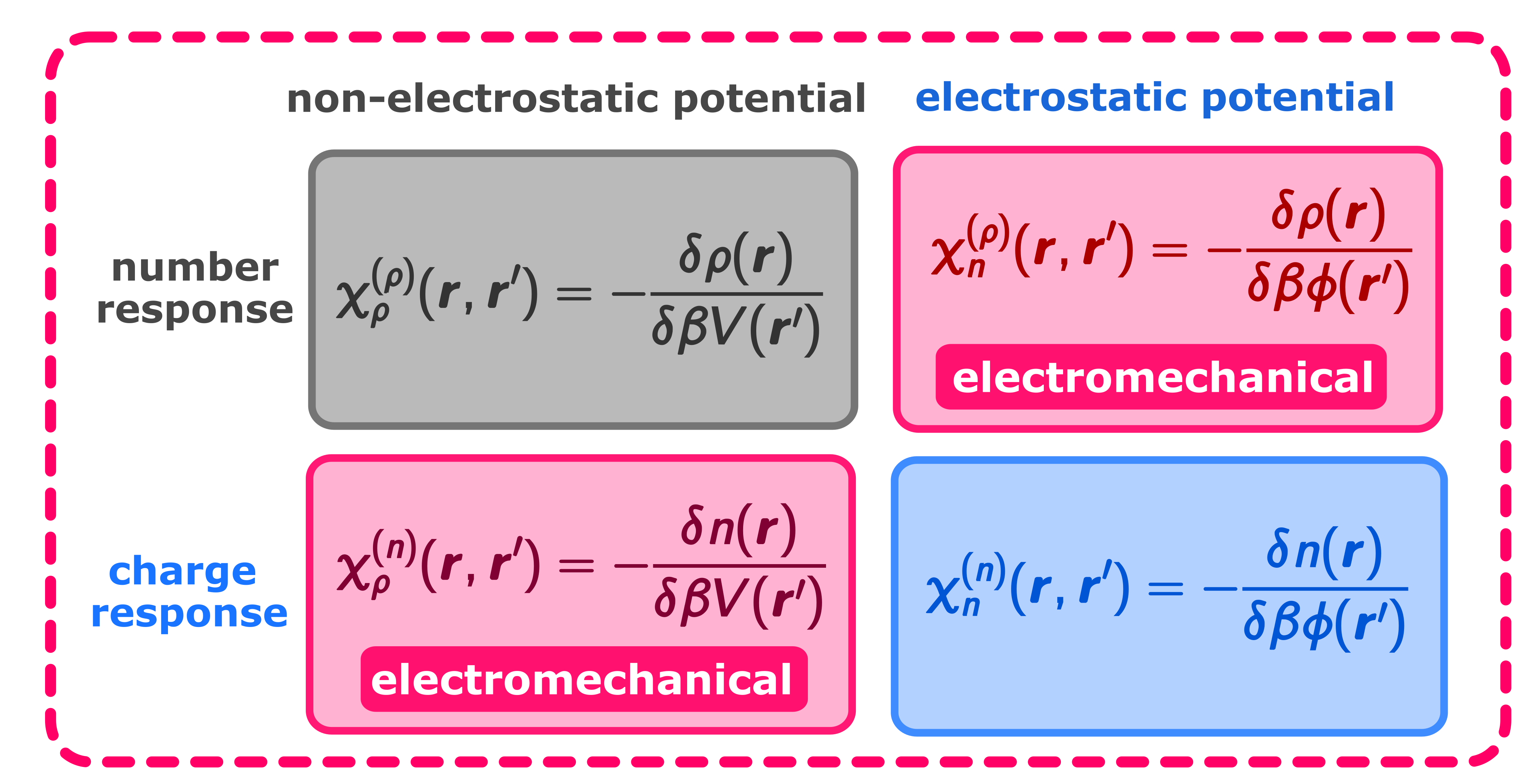 A first principles approach to electromechanics in liquidsAnna T. Bui, and Stephen J. CoxJ. Phys.: Condens. Matter, Jul 2025
A first principles approach to electromechanics in liquidsAnna T. Bui, and Stephen J. CoxJ. Phys.: Condens. Matter, Jul 2025Electromechanics in fluids describes the response of the number density to electric fields, and thus provides a powerful means by which to control the behavior of liquids. While continuum approaches have proven successful in describing electromechanical phenomena in macroscopic bodies, their use is questionable when relevant length scales become comparable to a system’s natural correlation lengths, as commonly occurs in, e.g., biological systems, nanopores, and microfluidics. Here, we present a first principles theory for electromechanical phenomena in fluids. Our approach is based on the recently proposed hyperdensity functional theory [Sammüller et al, Phys. Rev. Lett. 133, 098201 (2024)] in which we treat the charge density as an observable of the system, with the intrinsic Helmholtz free energy functional dependent upon both density and electrostatic potential. Expressions for the coupling between number and charge densities emerge naturally in this formalism, avoiding the need to construct density-dependent and spatially-varying material parameters such as the dielectric constant. Furthermore, we make our theory practical by deriving a connection between hyperdensity functional theory and local molecular field theory, which facilitates machine learning explicit representations for the free energy functionals of systems with short-ranged electrostatic interactions, with long-ranged effects accounted for in a well-controlled mean field fashion.
@article{bui2025principlesapproachelectromechanicsliquids, author = {Bui, Anna T. and Cox, Stephen J.}, title = {A first principles approach to electromechanics in liquids}, journal = {J. Phys.: Condens. Matter}, year = {2025}, month = jul, day = {8}, publisher = {IOP Publishing}, volume = {37}, pages = {285101}, doi = {10.1088/1361-648X/ade7e7}, url = {https://doi.org/10.1088/1361-648X/ade7e7}, }




2024
- AIP
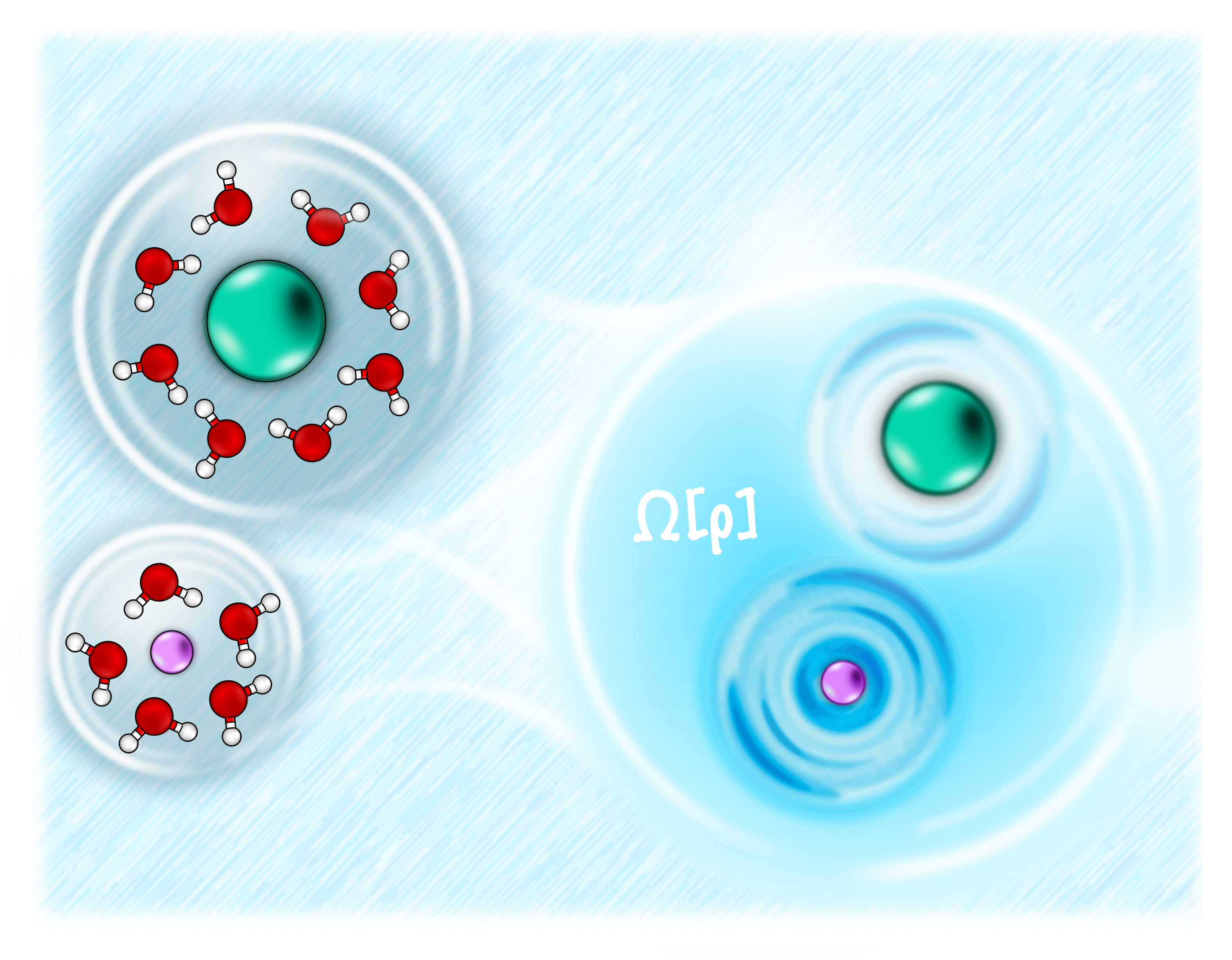 A classical density functional theory for solvation across length scalesAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Sep 2024
A classical density functional theory for solvation across length scalesAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Sep 2024A central aim of multiscale modeling is to use results from the Schrödinger equation to predict phenomenology on length scales that far exceed those of typical molecular correlations. In this work, we present a new approach rooted in classical density functional theory (cDFT) that allows us to accurately describe the solvation of apolar solutes across length scales. Our approach builds on the Lum–Chandler–Weeks (LCW) theory of hydrophobicity [K. Lum et al., J. Phys. Chem. B 103, 4570 (1999)] by constructing a free energy functional that uses a slowly varying component of the density field as a reference. From a practical viewpoint, the theory we present is numerically simpler and generalizes to solutes with soft-core repulsion more easily than LCW theory. Furthermore, by assessing the local compressibility and its critical scaling behavior, we demonstrate that our LCW-style cDFT approach contains the physics of critical drying, which has been emphasized as an essential aspect of hydrophobicity by recent theories. As our approach is parameterized on the two-body direct correlation function of the uniform fluid and the liquid–vapor surface tension, it straightforwardly captures the temperature dependence of solvation. Moreover, we use our theory to describe solvation at a first-principles level on length scales that vastly exceed what is accessible to molecular simulations.
@article{Bui2024solvation, author = {Bui, Anna T. and Cox, Stephen J.}, title = {A classical density functional theory for solvation across length scales}, journal = {J. Chem. Phys.}, year = {2024}, month = sep, day = {09}, volume = {161}, number = {10}, pages = {104103}, issn = {0021-9606}, doi = {10.1063/5.0223750}, url = {https://doi.org/10.1063/5.0223750}, } - AIP
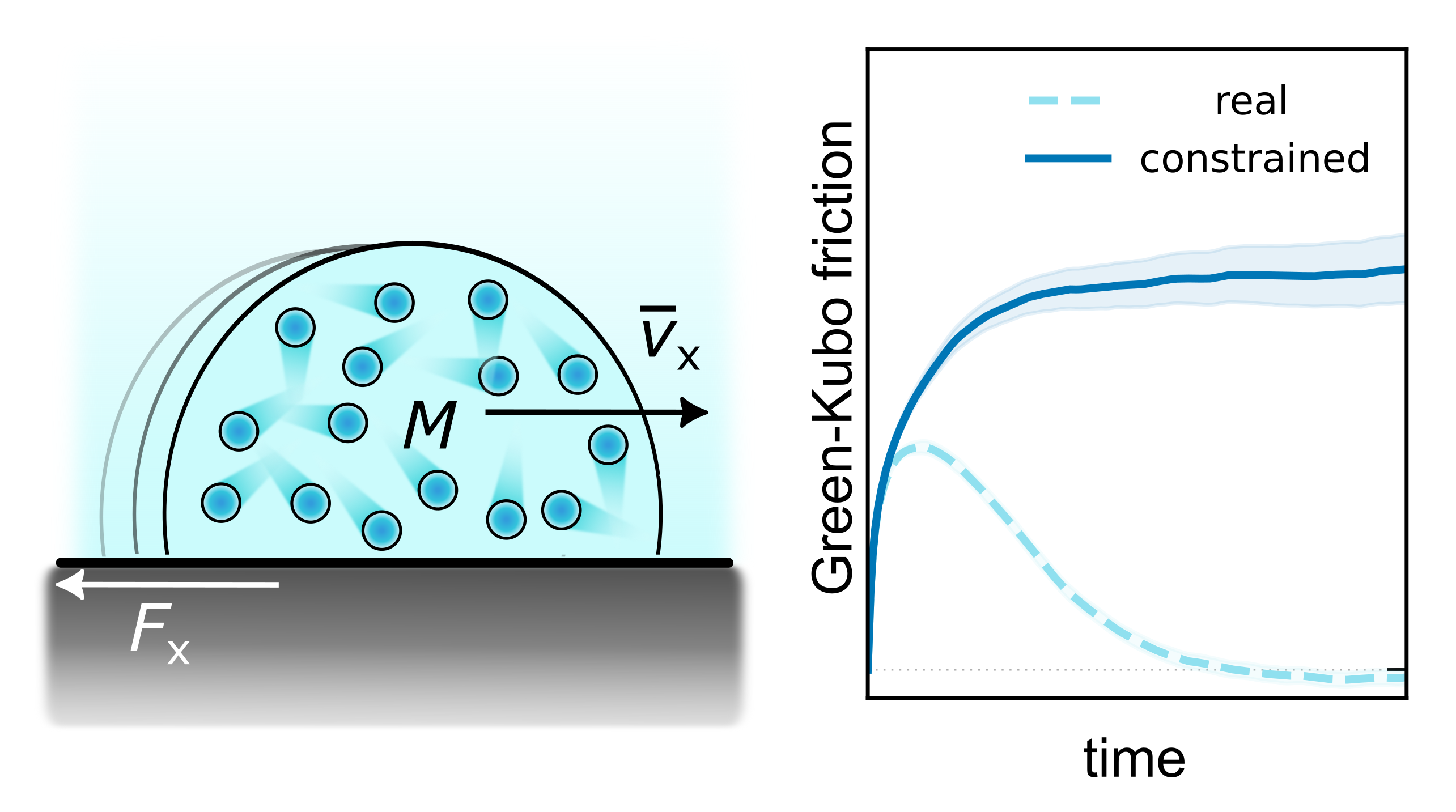 Revisiting the Green–Kubo relation for friction in nanofluidicsAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Nov 2024
Revisiting the Green–Kubo relation for friction in nanofluidicsAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Nov 2024A central aim of statistical mechanics is to establish connections between a system’s microscopic fluctuations and its macroscopic response to a perturbation. For non-equilibrium transport properties, this amounts to establishing Green–Kubo (GK) relationships. In hydrodynamics, relating such GK expressions for liquid–solid friction to macroscopic slip boundary conditions has remained a long-standing problem due to two challenges: (i) The GK running integral of the force autocorrelation function decays to zero rather than reaching a well-defined plateau value, and (ii) debates persist on whether such a transport coefficient measures an intrinsic interfacial friction or an effective friction in the system. Inspired by ideas from the coarse-graining community, we derive a GK relation for liquid–solid friction where the force autocorrelation is sampled with a constraint of momentum conservation in the liquid. Our expression does not suffer from the “plateau problem” and unambiguously measures an effective friction coefficient, in an analogous manner to Stokes’ law. We further establish a link between the derived friction coefficient and the hydrodynamic slip length, enabling a straightforward assessment of continuum hydrodynamics across length scales. We find that continuum hydrodynamics describes the simulation results quantitatively for confinement length scales all the way down to 1 nm. Our approach amounts to a straightforward modification to the present standard method of quantifying interfacial friction from molecular simulations, making possible a sensible comparison between surfaces of vastly different slippage.
@article{Bui2024gk, author = {Bui, Anna T. and Cox, Stephen J.}, title = {Revisiting the Green--Kubo relation for friction in nanofluidics}, journal = {J. Chem. Phys.}, year = {2024}, month = nov, day = {27}, volume = {161}, number = {20}, pages = {201102}, issn = {0021-9606}, doi = {10.1063/5.0238363}, url = {https://doi.org/10.1063/5.0238363}, }


2023
- ACS
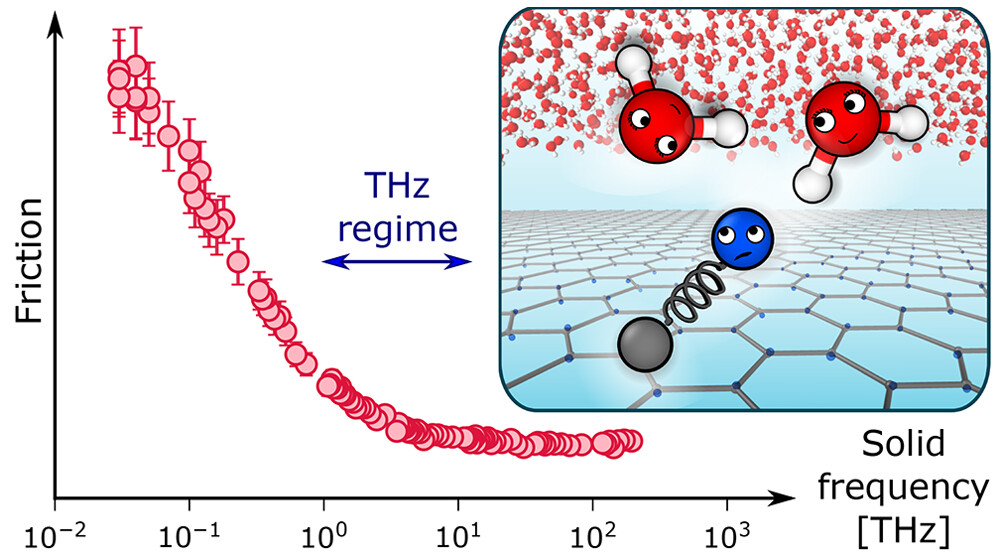 Classical Quantum Friction at Water–Carbon InterfacesAnna T. Bui, Fabian L. Thiemann, Angelos Michaelides, and Stephen J. CoxNano Lett., Jan 2023
Classical Quantum Friction at Water–Carbon InterfacesAnna T. Bui, Fabian L. Thiemann, Angelos Michaelides, and Stephen J. CoxNano Lett., Jan 2023Friction at water–carbon interfaces remains a major puzzle with theories and simulations unable to explain experimental trends in nanoscale waterflow. A recent theoretical framework—quantum friction (QF)—proposes to resolve these experimental observations by considering nonadiabatic coupling between dielectric fluctuations in water and graphitic surfaces. Here, using a classical model that enables fine-tuning of the solid’s dielectric spectrum, we provide evidence from simulations in general support of QF. In particular, as features in the solid’s dielectric spectrum begin to overlap with water’s librational and Debye modes, we find an increase in friction in line with that proposed by QF. At the microscopic level, we find that this contribution to friction manifests more distinctly in the dynamics of the solid’s charge density than that of water. Our findings suggest that experimental signatures of QF may be more pronounced in the solid’s response rather than liquid water’s.
@article{Bui2023qf, author = {Bui, Anna T. and Thiemann, Fabian L. and Michaelides, Angelos and Cox, Stephen J.}, title = {Classical Quantum Friction at Water--Carbon Interfaces}, journal = {Nano Lett.}, year = {2023}, month = jan, day = {25}, publisher = {American Chemical Society}, volume = {23}, number = {2}, pages = {580-587}, issn = {1530-6984}, doi = {10.1021/acs.nanolett.2c04187}, url = {https://doi.org/10.1021/acs.nanolett.2c04187}, }

2022
- ACS
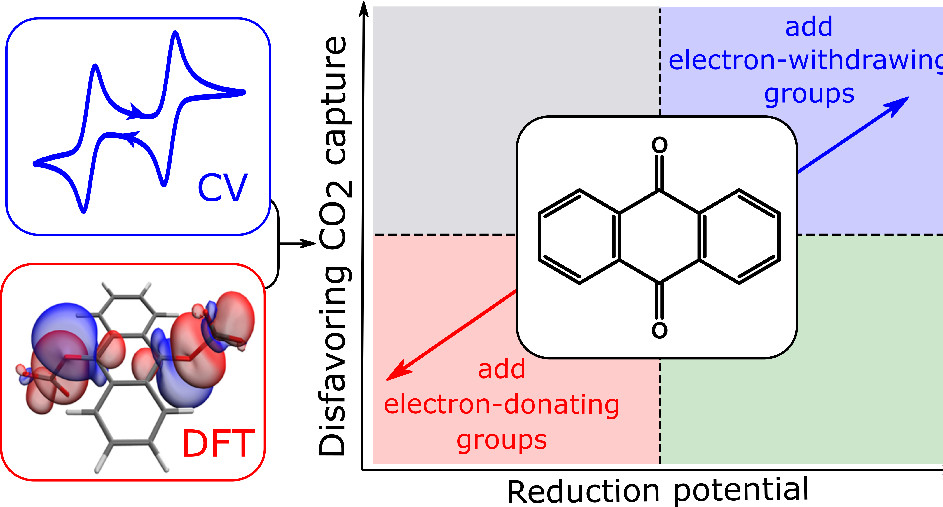 Trade-Off between Redox Potential and the Strength of Electrochemical CO2 Capture in QuinonesAnna T. Bui, Niamh A. Hartley, Alex J. W. Thom, and Alexander C. ForseJ. Phys. Chem. C, Aug 2022
Trade-Off between Redox Potential and the Strength of Electrochemical CO2 Capture in QuinonesAnna T. Bui, Niamh A. Hartley, Alex J. W. Thom, and Alexander C. ForseJ. Phys. Chem. C, Aug 2022Electrochemical carbon dioxide capture recently emerged as a promising alternative approach to conventional energy-intensive carbon-capture methods. A common electrochemical capture approach is to employ redox-active molecules such as quinones. Upon electrochemical reduction, quinones become activated for the capture of CO2 through a chemical reaction. A key disadvantage of this method is the possibility of side-reactions with oxygen, which is present in almost all gas mixtures of interest for carbon capture. This issue can potentially be mitigated by fine-tuning redox potentials through the introduction of electron-withdrawing groups on the quinone ring. In this article, we investigate the thermodynamics of the electron transfer and chemical steps of CO2 capture in different quinone derivatives with a range of substituents. By combining density functional theory calculations and cyclic voltammetry experiments, we support a previously described trade-off between the redox potential and the strength of CO2 capture. We show that redox potentials can readily be tuned to more positive values to impart stability to oxygen, but significant decreases in CO2 binding free energies are observed as a consequence. Our calculations support this effect for a large series of anthraquinones and benzoquinones. Different trade-off relationships were observed for the two classes of molecules. These trade-offs must be taken into consideration in the design of improved redox-active molecules for electrochemical CO2 capture.
@article{Bui2022, author = {Bui, Anna T. and Hartley, Niamh A. and Thom, Alex J. W. and Forse, Alexander C.}, title = {Trade-Off between Redox Potential and the Strength of Electrochemical CO2 Capture in Quinones}, journal = {J. Phys. Chem. C}, year = {2022}, month = aug, day = {25}, publisher = {American Chemical Society}, volume = {126}, number = {33}, pages = {14163-14172}, issn = {1932-7447}, doi = {10.1021/acs.jpcc.2c03752}, url = {https://doi.org/10.1021/acs.jpcc.2c03752}, }
