Anna T. Bui

My research uses statistical mechanics and computer simulations to model fluids’ behaviours that often spans across different length scales, capturing both microscopic correlations and mesoscopic phenomena. In my PhD, I focus on
- Equilibrium response: solvation phenomena, electric double layer
- Non-equilibrium response: how fluids flow under the nanoscale
My research heavily draws on theoretical techniques in soft matter (classical density functional theory, continuum hydrodynamics) and developing and applying them to systems of revelant in chemical physics (water, ions).
selected publications
- ACS
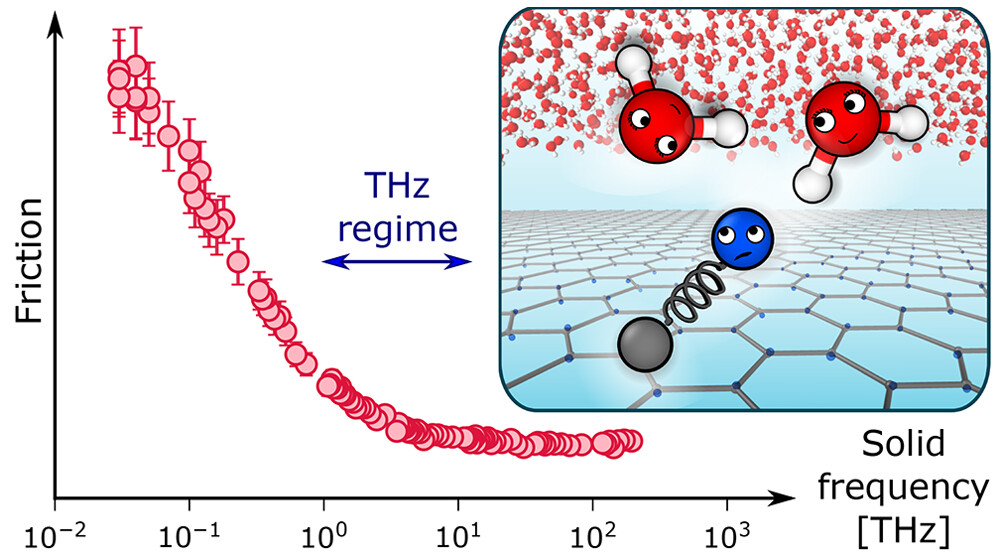 Classical Quantum Friction at Water–Carbon InterfacesAnna T. Bui, Fabian L. Thiemann, Angelos Michaelides, and Stephen J. CoxNano Lett., Jan 2023
Classical Quantum Friction at Water–Carbon InterfacesAnna T. Bui, Fabian L. Thiemann, Angelos Michaelides, and Stephen J. CoxNano Lett., Jan 2023Friction at water–carbon interfaces remains a major puzzle with theories and simulations unable to explain experimental trends in nanoscale waterflow. A recent theoretical framework—quantum friction (QF)—proposes to resolve these experimental observations by considering nonadiabatic coupling between dielectric fluctuations in water and graphitic surfaces. Here, using a classical model that enables fine-tuning of the solid’s dielectric spectrum, we provide evidence from simulations in general support of QF. In particular, as features in the solid’s dielectric spectrum begin to overlap with water’s librational and Debye modes, we find an increase in friction in line with that proposed by QF. At the microscopic level, we find that this contribution to friction manifests more distinctly in the dynamics of the solid’s charge density than that of water. Our findings suggest that experimental signatures of QF may be more pronounced in the solid’s response rather than liquid water’s.
@article{Bui2023qf, author = {Bui, Anna T. and Thiemann, Fabian L. and Michaelides, Angelos and Cox, Stephen J.}, title = {Classical Quantum Friction at Water--Carbon Interfaces}, journal = {Nano Lett.}, year = {2023}, month = jan, day = {25}, publisher = {American Chemical Society}, volume = {23}, number = {2}, pages = {580-587}, issn = {1530-6984}, doi = {10.1021/acs.nanolett.2c04187}, url = {https://doi.org/10.1021/acs.nanolett.2c04187}, } - AIP
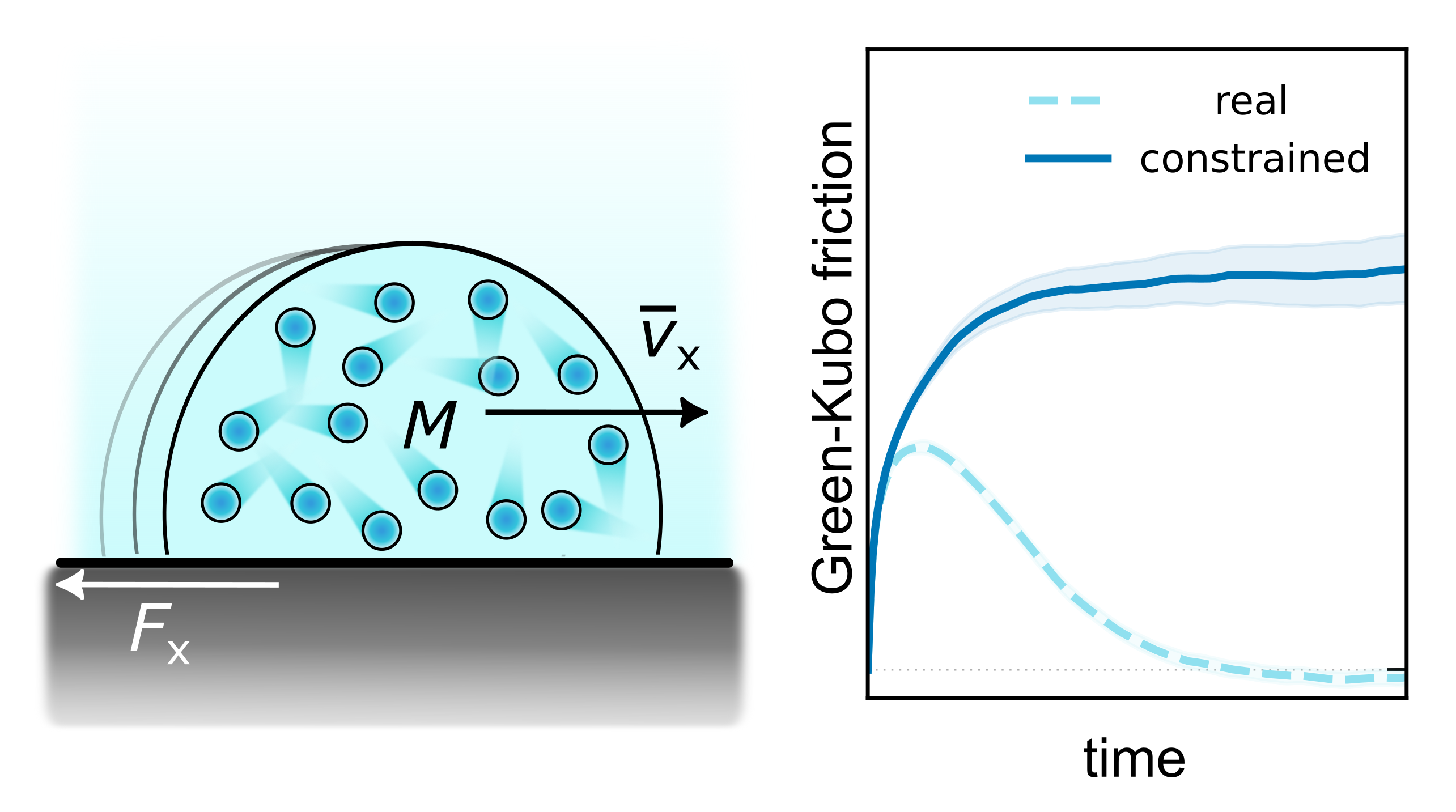 Revisiting the Green–Kubo relation for friction in nanofluidicsAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Nov 2024
Revisiting the Green–Kubo relation for friction in nanofluidicsAnna T. Bui, and Stephen J. CoxJ. Chem. Phys., Nov 2024A central aim of statistical mechanics is to establish connections between a system’s microscopic fluctuations and its macroscopic response to a perturbation. For non-equilibrium transport properties, this amounts to establishing Green–Kubo (GK) relationships. In hydrodynamics, relating such GK expressions for liquid–solid friction to macroscopic slip boundary conditions has remained a long-standing problem due to two challenges: (i) The GK running integral of the force autocorrelation function decays to zero rather than reaching a well-defined plateau value, and (ii) debates persist on whether such a transport coefficient measures an intrinsic interfacial friction or an effective friction in the system. Inspired by ideas from the coarse-graining community, we derive a GK relation for liquid–solid friction where the force autocorrelation is sampled with a constraint of momentum conservation in the liquid. Our expression does not suffer from the “plateau problem” and unambiguously measures an effective friction coefficient, in an analogous manner to Stokes’ law. We further establish a link between the derived friction coefficient and the hydrodynamic slip length, enabling a straightforward assessment of continuum hydrodynamics across length scales. We find that continuum hydrodynamics describes the simulation results quantitatively for confinement length scales all the way down to 1 nm. Our approach amounts to a straightforward modification to the present standard method of quantifying interfacial friction from molecular simulations, making possible a sensible comparison between surfaces of vastly different slippage.
@article{Bui2024gk, author = {Bui, Anna T. and Cox, Stephen J.}, title = {Revisiting the Green--Kubo relation for friction in nanofluidics}, journal = {J. Chem. Phys.}, year = {2024}, month = nov, day = {27}, volume = {161}, number = {20}, pages = {201102}, issn = {0021-9606}, doi = {10.1063/5.0238363}, url = {https://doi.org/10.1063/5.0238363}, } -
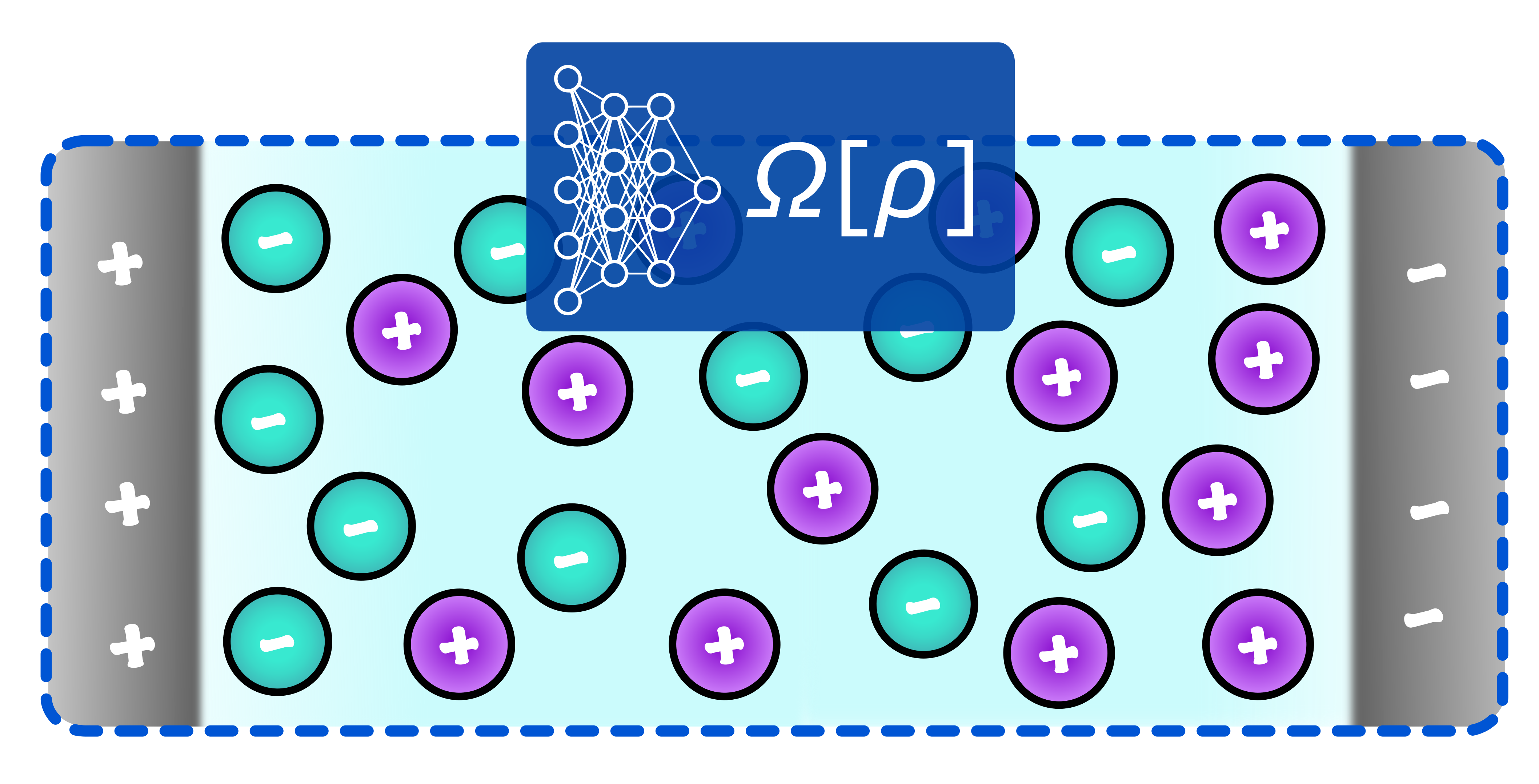 Learning classical density functionals for ionic fluidsAnna T. Bui, and Stephen J. CoxPhys. Rev. Lett., Apr 2025
Learning classical density functionals for ionic fluidsAnna T. Bui, and Stephen J. CoxPhys. Rev. Lett., Apr 2025Accurate and efficient theoretical techniques for describing ionic fluids are highly desirable for many applications across the physical, biological and materials sciences. With a rigorous statistical mechanical foundation, classical density functional theory (cDFT) is an appealing approach, but the competition between strong Coulombic interactions and steric repulsion limits the accuracy of current approximate functionals. Here, we extend a recently presented machine learning (ML) approach [Sammüller et al., Proc. Natl. Acad. Sci. USA, 120, e2312484120 (2023)] designed for systems with short-ranged interactions to ionic fluids. By adopting ideas from local molecular field theory, the framework we present amounts to using neural networks to learn the local relationship between the one-body direct correlation functions and inhomogeneous density profiles for a "mimic” short-ranged system, with effects of long-ranged interactions accounted for in a mean-field, yet well-controlled, manner. By comparing to results from molecular simulations, we show that our approach accurately describes the structure and thermodynamics of the prototypical model for electrolyte solutions and ionic liquids: the restricted primitive model. The framework we present acts as an important step toward extending ML approaches for cDFT to systems with accurate interatomic potentials.
@article{bui2025learningclassicaldensityfunctionals, author = {Bui, Anna T. and Cox, Stephen J.}, title = {Learning classical density functionals for ionic fluids}, journal = {Phys. Rev. Lett.}, volume = {134}, issue = {14}, pages = {148001}, numpages = {8}, year = {2025}, month = apr, publisher = {American Physical Society}, doi = {10.1103/PhysRevLett.134.148001}, url = {https://link.aps.org/doi/10.1103/PhysRevLett.134.148001}, } - arXiv
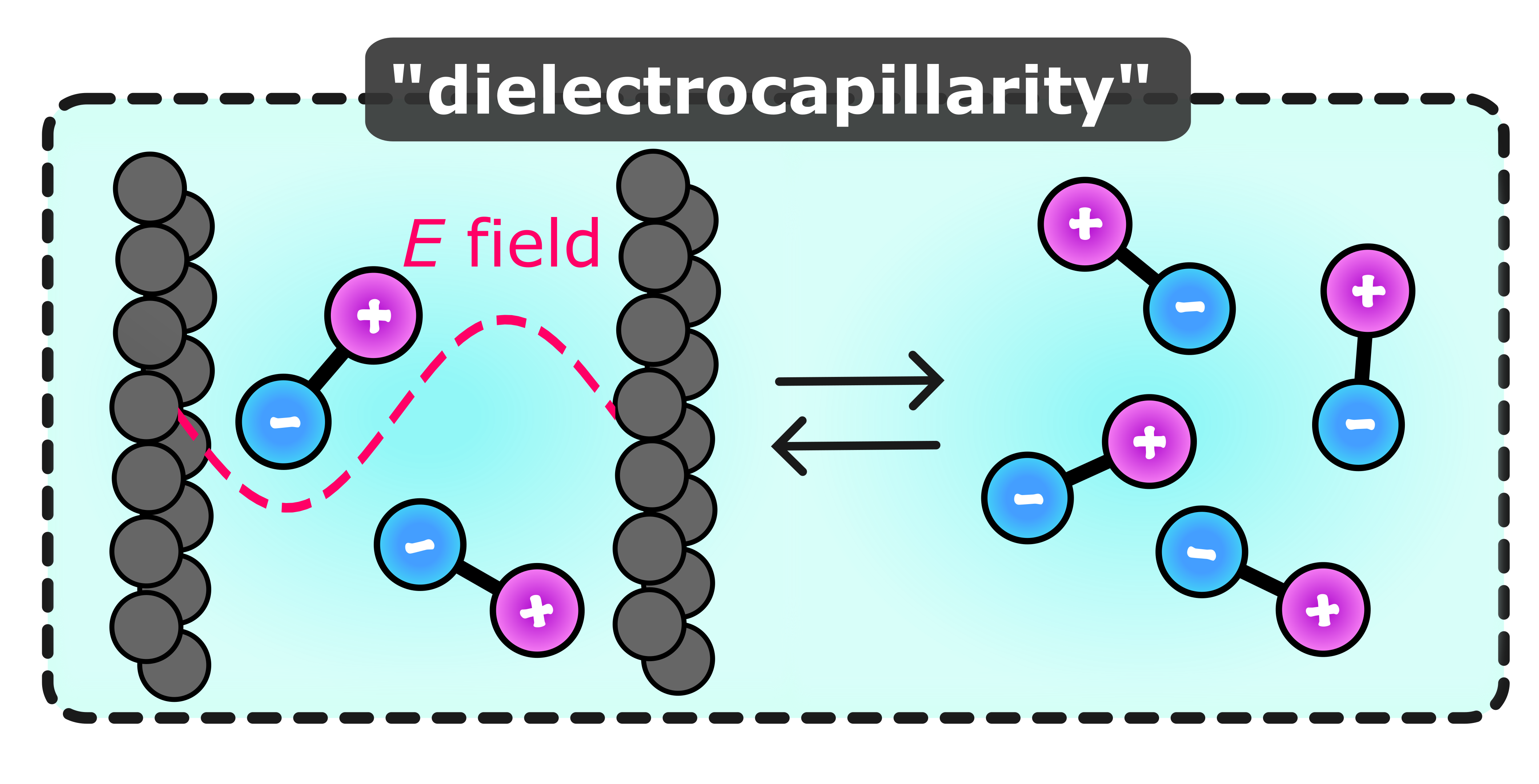 Dielectrocapillarity for exquisite control of fluidsAnna T. Bui, and Stephen J. CoxApr 2025
Dielectrocapillarity for exquisite control of fluidsAnna T. Bui, and Stephen J. CoxApr 2025Spatially varying electric fields are prevalent throughout nature and technology, arising from heterogeneity inherent to all physical systems. Inhomogeneous electric fields can originate naturally, such as in nanoporous materials and biological membranes, or be engineered, e.g., with patterned electrodes or layered van der Waals heterostructures. While uniform fields cause free ions to migrate, for polar fluids they simply act to reorient the constituent molecules. In contrast, electric field gradients (EFGs) induce a dielectrophoretic force, offering exquisite electrokinetic control of a fluid, even in the absence of free charge carriers. EFGs, therefore, offer vast potential for optimizing fluid behavior under confinement, such as in nanoporous electrodes, nanofluidic devices, and chemical separation materials. Yet, EFGs remain largely unexplored at the microscopic level owing to the absence of a rigorous, first principles theoretical treatment of electrostrictive effects. By integrating state-of-the-art advances in liquid state theory and deep learning, we reveal how EFGs modulate fluid structure and capillary phenomena. We demonstrate, from first principles, that dielectrophoretic coupling enables tunable control over the liquid-gas phase transition, capillary condensation, and fluid uptake into porous media. Our findings establish "dielectrocapillarity” – the use of EFGs to control confined fluids – as a powerful tool for controlling volumetric capacity in nanopores, which holds immense potential for optimizing energy storage in supercapacitors, selective gas separation, and tunable hysteresis in neuromorphic nanofluidic devices.



